■JPT-online■
| ◆ 第3章 本邦の臨床試験の質に関する分析と考察 | ◇ 第1節 | 目的 |
| ◇ 第2節 | 本邦の臨床試験におけるGCP違反・逸脱 | |
| ◇ 第3節 | GCP査察結果の日米比較 | |
| ◇ 第4節 | 発見された違反・逸脱の重大さ | |
| ◇ 第5節 | 新GCPの実施と本邦における臨床試験の質の変化 | |
| ◇ 第6節 | 新GCP下で実施された試験のGCP調査結果 | |
| ◆ 文 献 | ||
| ◆ 表 | ||
 へ へ |
||
 へ へ |
||
第2節 本邦の臨床試験におけるGCP違反・逸脱
Table 3-1に 医薬品機構が1997年4月から2000年3月の間に医療機関を訪問して実施したGCP調査の実施状況を示す 24)24)Ono S, Kodama Y, Nagao T, Toyoshima S. The quality of conduct in Japanese clinical trials: deficiencies found in GCP inspections. Controlled Clinical Trial (in press).(なお,参考までに2000年4月から1年間の数字も付す)。
この観察時期に,医薬品機構は延べ331の医療機関の調査を実施した。医療機関のタイプでみると, 私立病院が155施設と最も多く,公立病院63施設,私立大学病院52施設と続く。この331調査は775プロトコルを対象に行われた。 つまり,平均すると,医薬品機構は一つの医療機関を訪問して(通常,調査は1日),2.3プロトコルの試験実施状況を調査したことになる。 また,調査対象品目数は125(成分数ベース)であった。
Table 3-1の調査により発見されたGCP違反・逸脱の内容がTable 3-2である。Table 3-2の件数は, 医薬品機構の調査報告書における1つの指摘を1件と数える。たとえば,「プロトコル番号6の症例番号5の血圧の記載に誤りがあった」, 「プロトコル番号8に関する治験審査委員会の審議の議事録が作成されていなかった」等はすべて1件と数えられる。 指摘はGCPの条文単位・症例単位で行われることが多いが,いくつかの症例についてまとまった記載となることもある。
Table 3-2の詳細な分析に入る前に, 今回の調査結果の解釈,特に一般化可能性(この分析が日本の試験全体に当てはまるか)についての制約・限界について述べる。
第一に,今回の調査は厚生労働省に承認申請があった医薬品の臨床試験に対するものである。 つまり,これらのほとんどは予期した目的が達成された試験である。 期待した成績が得られなかった試験の質は,総体的にこれよりも低い可能性があるが, これについての知見は今のところ入手不可能である。また,調査を行う医療施設はランダムには抽出されていない。 抽出には政策的意図が働いており,本邦の医療機関を万遍なくカバーするものではない。
第二に,Table 3-2に示す件数がそのまま試験の質の指標とはなりえない。 違反発見件数は,実際の違反発生状況のみならず,GCP調査を行なう担当者の問題意識,規定条文の詳細さにも依存する。
第三に,個々の違反・逸脱の程度(重大さ)は, Table 3-2からは 読み取ることができない。先述のとおり,非常に深刻なGCP違反が生じた場合には,申請資料からその臨床試験成績が削除されることになるが, Table 3-2の 違反のほとんどはそのような深刻なものではない。なお,違反の重大さ・深刻さについては第4節で分析する。
第1項 試験実施医療機関が関係する違反・逸脱
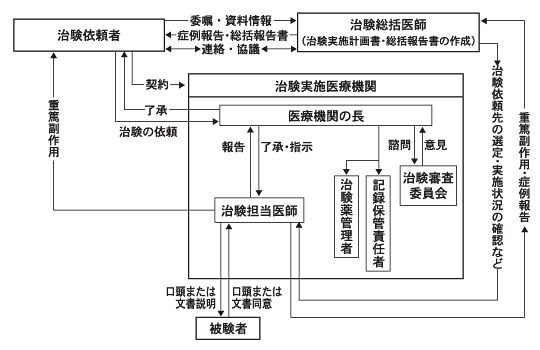
Table 3-3に試験を実施する医療機関に関係する違反・逸脱を示す。 本邦の試験の契約は,医療機関の長(病院長)と製薬企業の間で締結される(Fig.1-1, 1-2, 「わが国の臨床試験とそれを評価するための枠組み-1 第1章 序論─本邦の臨床試験の仕組み─」を参照)。 つまり,病院における試験実施の最終的な責任は,その意味では病院長にある。治験審査委員会の設置も病院長の義務である。
Table 3-3では,試験契約に関係する逸脱が多く見られる(d-1,d-2)。 これらの逸脱は,試験結果の質に悪影響を与えるものは少なく,むしろ, 日本の予算制度の融通のなさ,手続き的な非効率に由来するものが多いようにも思われる。 医療機関が試験を実施する適格性を有するか(a)は,スタッフや設備の観点で不十分な施設で試験が実施されたとの指摘である。 たとえば,パートタイム勤務の医師を試験担当医師として,当該医師が到底遵守不可能なプロトコルを医療機関が契約することも本項の違反となる。
治験審査委員会(IRB)に関して観察された違反には,旧GCPから新GCPへの移行期であることに由来するものも含まれる。 なお,対象となる試験の実施時期は旧GCP下であっても,IRBに関しては,新GCPの施行に合わせて, GCP調査の時点で行われている試験にも規定が適用されている点に注意が必要である。 たとえば,IRBの標準業務手順書(SOP)に関する指摘(b-2)は,そのようなSOP文書が本邦では初めて導入されたことを考えると, 移行期の準備不足の反映とも言えそうである。IRBの委員構成が不適格(c-1)という点も同様である。
IRBでの審議が不十分という点(c-2)は昔から指摘されている 28)28)Fukuhara S, Tanabe N, Kurokawa K. Background issues in Japan affecting the ethical review process for clinical trials. Int J Pharmaceut Med 1999;13:25-8.。 医療機関があまりにも多くの試験を引き受けていることがその主たる原因である 29)29)上田慶二, 福原俊一, Glickman P, 田邊昇, 黒川清. 新GCP導入がわが国の臨床試験に与えた影響と問題に関する調査. 臨床評価 1999; 27: 371-38.。 IRBの議事録が不十分であること(c-4)も数多く指摘されているが,IRB事務局が充実するにつれてこのような指摘は減ることが予想される。 実施中の試験の継続的なフォローアップをIRBが怠っているケースも多かった(c-3)。
Fig.1-1 新GCP下の治験実施体制
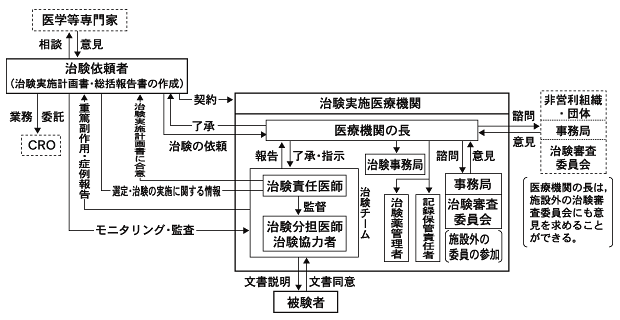
Fig.1-2 旧GCP下の治験実施体制
第2項 治験実施医師が関係する違反・逸脱
Table 3-4に 試験を実施する医師に関係する違反・逸脱を挙げた。もちろん,これらの逸脱すべてについて治験実施医師に主たる責任があるわけではなく, 被験者側の要因やプロトコル自体の欠陥に起因する逸脱も多い。
多く見られる登録基準違反(e-1)としては,年齢違反,既往歴に関する逸脱から, 倫理的見地から格段の配慮が必要な被験者の登録に関する違反(たとえばアルツハイマー病患者,自社製品の試験に参加する企業社員等)が挙げられる。
プロトコルで使用が禁止されている併用薬を使用すること(e-2)は試験の質を著しく低下させるが, 本邦ではこのような違反がきわめて多く発生する。これは,日本の臨床試験が, 基本的にあらゆる薬剤を処方可能な国民皆保険の医療制度下に組み込まれていること, 日本の医師が,一般的に,多剤処方を好むことによると思われる30)30)Ikegami N, Mitchell W, Penner-Hahn J. Pharmaceutical prices, quantities and innovation. Comparing Japan with the US. Pharmacoeconomics 1994; 6: 424-33.。 また,臨床検査がプロトコルどおりに実施されない点(e-3)も,試験データの質および被験者の安全確保の観点から問題である。
Table 3-2およびTable 3-4からわかるとおり, 指摘の中で最も多いのが症例報告書(case report form。以下「CRF」とする。)の記載内容が不正確というものである。 その中で最も多いのが実際に使用された併用薬等の不記載(f-1)である。 先述の多剤処方傾向が背景にあるのはもちろんであるが, 加えて,「薬効に影響を与えない薬剤の投与はCRFに記載する必要がない」という誤解を有する医師がかつては多数いたことも事実である。 たとえば,胃粘膜保護剤等の記載漏れは多くの場合この誤解に基づいていた。いうまでもなく,このような投薬は安全性評価上の重大なバイアスとなる可能性がある。
臨床検査値(f-2)や有害事象の誤記載(f-3)は,医師の不注意が直接の理由ではある。 しかし,こうしたCRFの記載ミスを単に医師の不注意のみに帰することは明らかに誤りである。 真の原因は,本邦の臨床試験に見られた次の三つの決定的な欠陥である。
第一に,旧GCP下では,治験依頼者たる企業の開発担当者が,医療機関の原データ(カルテ等に記入されている生データ)を閲覧し, 試験が実際に適切に行われていることを確認することが許されていなかった。 正確には,これはGCPではなく薬事法の守秘義務に関する欠陥であったが,この欠陥は1996年の薬事法改正で修正されている。
第二に,本邦の試験の現場には,欧米でresearch nurseと呼ばれる試験実施医師をサポートする職種が存在しなかった。 本邦においてresearch nurse,clinical research coordinator(CRC)と呼ばれる治験協力者が広く活動を始めるのは,新GCPの実施以降である。
第三に,上記二つの欠陥の結果として,医師が正確に,かつ,適切なタイミングでCRFを作成し,提出するインセンティブが存在しなかった。 CRFは一覧表形式のものが主流で,医師は試験終了時に実施した症例のCRFをまとめて作成することも多かった。
この三つのうち,第一の欠陥が第二,第三の欠陥を生み出す背景となっていたという意味で,最も重大であったと考える。 新GCPにおいてモニタリング・監査の実施の義務が依頼者に,また,その受け入れ義務が医療機関に課されたことにより, 第二,第三の欠陥もその対応の一環として次々に改善されている事実がそれを裏付ける。
第二の欠陥に関連して,日本ではチーム医療の概念が歴史的に欠如していた点も知られている 31)31)Tominaga T. Japan's New GCP regulations and their impact on clinical trials. In: Tominaga T, ed. Japan's new GCP and other rules on clinical trials (Part 3). Tokyo: Yakuji Nippo; 1999.。 また,従来は,原因・結果の議論は別にして,試験から得られる金銭的メリットの多くが医師(医局)に向かい, 看護婦や薬剤師の試験関連業務に対して適切な報酬が支払われるメカニズムがなかったことも事実である。
なお,医療機関の側から治験実施医師に係る問題点に対応するためには, さらに,大学・医療機関の教育・研修過程における臨床研究全般(臨床試験を含む)についての教育の充実が必要である。 特に,臨床経験等が比較的浅い医師が,試験責任医師の指導・監督の下,試験分担医師として初めて試験に参加するような場合には, 臨床試験の科学(方法論)・倫理の十分な理解を得るための何らかの研修が必要であり,また,有用であろう。
本邦におけるCRFの不正確さが具体的にどのような帰結をもたらすかについては, 有効性に関する結論を期待する試験における分析感度assay sensitivityの観点からの議論が可能である 32)32)Temple R, Ellenberg SS. Placebo-controlled trials and active-control trials in the evaluation of new treatment. Ann Intern Med 2000; 133: 455-63.。 プラセボの使用が歴史的に好まれない日本では,有効性の検証目的の試験として同等性試験(厳密には非劣性試験)が実施されることが多かった。 そのような試験で,CRFの作成が不正確に行われているとすれば,それは試験の分析感度を失わせる方向に作用し, 被験薬が有効であるとの結論を疑わせることとなる33)33)International Conference on Harmonisation (ICH) Harmonised Tripartite Guideline. Choice of control group and related issues in clinical trials. Geneva: ICH-secretariat, 2000.。
プラセボの使用を好まないという被験者・医師の傾向自体が当面変わらないとすれば, CRFの不正確さは薬効評価における重要な問題として今後も強く監視されるべき事項である。
第3項 インフォームドコンセントに関係する違反・逸脱
患者からインフォームドコンセントを得る際の違反・逸脱を Table 3-5に示す。
Table 3-5の 指摘のほとんどは,重大な倫理的な問題点が見出されたというものではなく,手続き上の逸脱であった。 第3節において述べるとおり,日本のインフォームドコンセント関係の違反の数は, 米国のそれと比較してかなり少ないように思われるが3434)Shapiro MF, Charrow RP. Scientific misconduct in investigational drug trials. N Engl J Med 1985; 312: 731-6., 35)35)Weiss RB, Vogelzang NJ, Peterson BA, Panasci LC, Carpenter JT, Gavigan M, Sartell K, Frei E, Mclntyre OR. A successful system of scientific data audits for clinical trials: a report from the Cancer and Leukemia Group B. JAMA 1993; 270: 459-64., そのことは必ずしも本邦の試験に倫理面の問題がないことを示すものではない。 たとえば,老人養護施設のアルツハイマー病の患者を適切な同意なしに試験に参加させたことが 判明した事件等が最近の例として挙げられる36)36)朝日新聞, 1997 Jan 22:30.。
小児の試験等で適切な代諾者の同意が得られていなかったケース(g-1), 同意の際に説明が義務付けられている「治験に参加しない場合の治療上の選択肢」が十分に説明文書に盛り込まれていないこと(g-2)等の逸脱も見られている。 これらも,わが国の一般医療の特徴を反映したものと考えられる。
第4項 治験薬の管理,資料保管に関する違反・逸脱
新GCP下の試験だけでなく,旧GCP下においても,医師が直接治験薬を管理する場合を除いて,病院の薬剤部(科)が試験薬の管理を行うことが多かった。 また,薬剤部は,多くの場合,事実上の臨床試験の事務局を兼ねていることも多く,その働きが試験の円滑な実施の鍵を握っていた。
Table 3-6に 薬剤の取り扱いと資料保管に関する違反・逸脱の件数を示した。
不適切な薬剤管理(h-1)の例としては,試験中に薬剤が不適切な場所(たとえば医師の個人的な机)に保管されていたり, 紛失したりするケースが挙げられる。治験薬管理票の作成が行われていないことも多く,特に医師が自ら治験薬を管理していた医療機関では, 治験薬の出納の記録が全く残っていないこともあった(h-2)。
原資料(カルテ,検査結果,説明同意文書等)の保管・管理にも問題が見られた。 GCP調査に訪問した医療機関で,原資料の根幹となるカルテが廃棄されて存在しないことが時としてあった(i-1)。 これは医療法で定められたカルテの保管義務(5年間)を超える期間の保管について, 臨床試験の資料保管を定める薬事法に明確な定めがなかったことが主たる原因である。この矛盾も新GCPの策定とともに修正された。
カルテに挟んであったインフォームドコンセント文書が紛失してしまったというミスも散見された(i-2)。
こうした違反を減らすためには,多忙な医師に文書管理のすべての責任を負わせるのではなく,CRCや治験事務局が責任の一部を分担する仕組みが必要であろう。