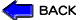■JPT-online■

はじめに
臨床試験の基盤整備について,がん臨床試験の立場から問題点を指摘し,その在り方と整備の必要性を提言したい。
わが国におけるGCPの要件と問題点
わが国では臨床試験は二つに分かれている。表1に示すように一つは企業の行う新薬開発治験で,新GCP(good clinical practice)による法的行政的規制がかかっている1)。もう一つは研究者が公的研究費を得て行う公費臨床試験で, 患者に対する最善の治療法や標準的治療法,証拠に基づいた医療(EBM: evidence-based medicine)を確立するために必要な証拠(evidence)を創ることを目的にして行われる。 がんの領域では特にこれが必要である。しかしこれに対して新GCPの規制は何らかけられていない。 規制もかけない代わりに,公的研究費の支援もごくわずかしかしないというのが現状である。
| 高血圧 | MARHY STOP,SHEP,HOT |
βブロッカー+利尿薬 高齢者高血圧 Jカーブ現象 |
|---|---|---|
| 心不全 | PROMISE CONSENSUS US-Cardilol DIG |
PDE阻害薬(強心薬) ACE阻害薬 βブロッカー ジギタリス |
| 虚血性心疾患 | BHAT SAVE TIMI BENESENT |
βブロッカー ACE阻害薬 t-PA ステント |
| 不整脈 | CAST STAT-CHT |
I群不整脈薬 アミオダロン |
| 高脂血症 | WOSCOPS4S, CARE | 1次予防 2次予防 |
この二つの臨床試験の形態について,もう少し説明しておく必要がある。進行期悪性腫瘍の薬物療法を成功させるには,まず優れた新規抗がん剤の臨床開発治験が必要である。 治験は製薬企業主導型の臨床研究である。この治験の研究成果に基づいて,新薬の製造承認,保険適応,市販が厚生省より認可される。 しかし,市販される優れた新薬といえども,単剤でがんを治すことはできない。そこでがんの治癒を目的として優れた多剤併用療法の臨床開発とそれを標準的治療法として確立するために公費臨床試験研究を行うことが必要となる。 これは製薬企業ではなく主として臨床研究医師主導型の公費臨床試験により行われる。この研究により標準的治療法が確立するとがんの治癒率が向上し,がん患者に対し最善の医療が提供できるようになり,医療の質も向上する。 すなわち,企業主導型の新薬開発治験と臨床研究医師主導型の標準的治療法確立のための公費臨床試験の両者はがん薬物療法を成功させるために,ひいてはがん患者に対し治癒が期待される最善の治療法を提供するために必須な臨床研究である。
さて,患者を対象とする治療研究は,表1に示すように倫理性,科学性,信頼性を確保して行う必要がある。 そのため1989年から日・米・ヨーロッパ連合三極の新医薬品承認審査資料の規制に関する国際整合化会議(ICH: International Conference of Harmonization)が行われ,新医薬品の品質,安全性,有効性に関する数多くのICHガイドラインが定められた。 各国はこれを批准して国内に導入し,その法的,行政的規制下で臨床試験を行うことが要請されている。 このうち,有効性に関する臨床試験の規制がGCP(good clinical practice)と呼ばれるものである。 わが国では1998年から新GCPとして導入されたが,この規制を受けるのは製薬企業主導型の新薬開発治験のみであり,公費臨床試験はその枠外に置かれている。 これは全く片手落ちな研究政策であり,わが国の特殊な状況といってもよい。このような体質が米国とわが国の臨床研究体制や臨床試験の質に大きな差をもたらしている。
米国では治験と公費臨床試験は,両者とも国家研究法の規制下にあり,GCPによる法的,行政的規制を受けている。 倫理面では1960年代の早くからインフォームド・コンセントや機関審査委員会(IRB: institutional review board)の役割が明確に定められ,臨床試験はその科学性,倫理性,信頼性を一定レベル以上に保つことが要求されている。
臨床試験の科学性と信頼性は多施設共同研究グループ機構と臨床研究体制があってはじめて確保される。米国の共同研究グループ機構は公費臨床試験を通して構築されてきた。 そのための公的研究費は,臨床試験研究の保健医療的,社会的役割の重要性を考慮して,政府よりそれなりに十分支給されている。 臨床試験研究に規制をかけるが,それなりの研究費も与えているのである。一方,わが国では公費臨床試験に対し規制をかけないが,公的研究費の支援もあまりしないという研究政策がとられている。 公的研究費が乏しいため多施設共同研究に必要な共同研究グループ機構(臨床研究グループ,データセンター,各種委員会)は構築されてこなかったし, 各施設の臨床研究体制に必要な臨床研究医師の資格付け,臨床疫学や生物統計学の教育講座,病院にリサーチナースやデータマネージャーなどの臨床研究コーディネーター(CRC: clinical research coordinator)の定員化,臨床研究に必要な研究設備などの整備も当然行われていない。 このため,わが国では患者のための標準的治療法確立に必要な公費臨床試験研究は活力がない状況に置かれている。 したがって標準的治療法のほとんどは欧米で行われた公費臨床試験により確立され,証拠に基づく医療EBMのほとんどは米国産という残念な結果になっている。 日本では治療研究に対する国の研究政策そのものが不十分で,わが国独自の臨床研究結果に基づいて医療の質の向上を図ることは困難な状況にある。これは早急に改善すべきことである。
1 倫理性
治験および公費臨床試験における倫理の問題はヘルシンキ宣言がもとになっている。治験はインフォームド・コンセントを文書で行い,その内容および文書一式はIRBが審査することが新GCPで義務づけられている。 治験の場合,IRBは治験審査委員会と呼ばれている。公費臨床試験の場合もIRBないしは倫理委員会(IEC: independent ethical committee)が審査し,許可された方法に従ってインフォームド・コンセントを得て研究を行うことが国際的なルールである。 しかし,わが国の公費臨床試験ではこれは必ずしも義務づけられていない。このことは問題である。
2 科学性
治験の科学性は新GCPが導入されたことによりかなり改善された。研究の科学性を保つため,プロトコール,症例記録票(CRF: case report form)および臨床安全性情報を用意し,それに従って治験を行うことが求められている。 厚生省は治験に対し,新GCPは省令で,新GCP関係ガイドおよび薬効別の臨床評価ガイドラインは通知で出し,それによって科学性の基本レベルが確保されるように制度化した。 すなわち治験ではこれらのガイドライン,プロトコール,インフォームド・コンセント文書を遵守して研究を行うことが要請されている。 治験を科学的に適切に,かつ効率よく行うため,最近では企業は治験のプロトコール研究管理を開発業務受託機関(CRO: contract research organization)に依頼することも行われている。 一方,参加施設は各自の臨床研究体制を構築して治験を行う必要があり,そのために必要となる研究事務やデータ整理を行う人員をSMO( site management organization) から派遣してもらうことも可能になりつつある。
わが国では,公費臨床試験の科学性を保つためには,国際的なICH-GCPや各種のICHガイドライン,ならびに薬効別の臨床評価ガイドラインなどを遵守して行う必要があるが, 米国のような国家研究法による規制は全くかけられていない。すべて研究者が自主的に規制して行うことになるため,その研究の質やその科学性はピンからキリまで生じることになる。 患者に対する最善の治療法や標準的治療法を確立するために,必須となる公費臨床試験が制度化されていないことは,国の研究政策,行政の認識,大学医学部の研究評価,研究病院の臨床研究体制に問題があることを示している。 このなかでも,国の研究政策の影響が絶大である。
一方,公費臨床試験を行うには,共同研究グループが最初から形成されており,その中にはデータセンターや各種委員会組織などの共同研究グループ機構が必要である。 しかしわが国ではこの機構を備えている共同研究グループはきわめて少ない。欧米ではむしろこの共同研究グループ機構が先に作られ,そこで公費臨床試験も治験もともに行われている。 また,共同研究グループの本部は公益財団になっており,そこでCRO的な研究業務も行うことができるようになっている。
また,わが国の公費臨床試験を行う施設の臨床研究体制もきわめて不十分といわざるを得ない。CRCはわが国ではほとんど養成されてこなかったし,その定員化もはかられてこなかった。 やっと平成11年度より3国立センター病院と3国立大学病院に対し各1名ずつのCRCの定員がついた所である。 すなわち,これまでは公費臨床試験に必要な共同研究グループ機構や臨床研究を行う施設内の臨床研究体制が構築されてこなかったため,わが国では治験や公費臨床試験を適正に実施することが困難な状況にある。
臨床試験の研究業務を分担して行う職種を一般的にわが国ではCRC,米国ではCRA(clinical research associate)と呼び,その中に,リサーチナース(research nurse), 研究コーディネーター(SC: study coordinator)またはCRC,薬剤師(pharmacist),データマネージャー(DM: data manager)またはデータコーディネーター(DC: data coordinator)などの専門職がある。 これらの職種の方々を雇用する場合,わが国では施設が国公立であるか,私立であるかによって大きな差が生じている。 私立の場合,治験の受託研究費は収益とみなされ,課税される。しかし,施設はこの収益により,その資金でCRCを定員で雇用できることになる。 一方,施設が国公立の場合では定員法の枠があるため,受託研究費が入っても,CRCは定員で雇用できず,すべて非常勤職員になる。 これでは身分が非常に不安定となる。さらに,公費臨床試験の場合では,公的研究費といえども,その取扱規程により規制がかけられており,アルバイト雇用しか認められていない。 そのため処遇はいっそう悪くなり,能力のある人はほとんどなり手がない。 国立では私立のように受託研究費は課税の対象になっていないが,技術料,機械損料,建物使用料などの名目で研究費の30%を上乗せしており,これはそのまま国庫へ納入させている2)。 つまり,受託研究費に対し,国が30%の使用料的税金をかけているといわれても仕方のない状況である。これはむしろ米国のように施設の研究体制の整備に使うべきものである。 このような状況のため,臨床研究体制は非常にお粗末な状況にある。
以上より治験や公費臨床試験について,その研究の質やその科学性を確保することは,わが国では制度的に困難な状況にある。しかし,これは早急に改善しなければならないものである。
3 信頼性
治験および公費臨床試験の信頼性は,品質管理,品質保証,査察(実地調査)という研究業務によって確保される。 研究の品質は一般的にモニタリングを行うことで確保されるが,治験の場合は企業のモニターにより,公費臨床試験ではデータセンターのモニターにより行われる。 品質保証はモニタリングなどによる品質管理が新GCP基準,ガイドライン,プロトコールを遵守して行われているかどうかを監査するものである。 治験の場合は企業の監査員が,公費臨床試験の場合は公的研究費のスポンサーが監査を行う。この監査はかなり教育的に行うのが通例である。 監査で大きな問題点が指摘されれば,規制当局が査察(実地調整)に入ることになる。この規制当局が行う査察は本来罰則規程のある厳しいものであるが,わが国ではとてもそこまで厳しくできない状況にある。 わが国の臨床試験研究が今まで制度化されていなかったこともあって,この信頼性を確保するための研究業務が全く行われてこなかったし,その重要性がなかなか認識されていないのは問題である。
4 研究結果の行政への反映
治験ではその研究成果を行政に申請すれば,新薬の製造,保険適応,市販などが厚生省で承認されるという道はあるが,公費臨床試験では標準的治療法や適応拡大の有用性が確立しても, わが国ではそれを行政が取り上げることはないという状況が長く続いてきた。やっと最近になり,適応拡大については,公費臨床試験の成果が論文で公表されたあと, それを薬事行政に取り込めるようにする通知3)が出され,適応拡大の承認は平成11年の暮れより少しずつ実行されてきた。 しかし,実際には欧米で適応が承認されているが,わが国では適応が承認されていない医薬品について,その適応拡大承認を目的の一つとして公費臨床試験を行う場合, 保険の査定をうけたり,事故が起こった時の補償をどうするかなどの問題が残っている。厚生省からはこのような点も含めて実行できる通知として出してほしい。
新薬および標準的治療法の臨床開発とその確立に関する臨床研究の歴史的展開:
白血病や悪性リンパ腫の薬物療法を例として
白血病や悪性リンパ腫の薬物療法を例として
米国ではいち早く戦前から抗がん剤の臨床開発研究が行われ,戦後15年ぐらいの間に白血病に多くの有効な抗がん剤が開発されてきた。 この研究をリードしてきた米国での臨床試験研究の歴史的経過を知ることは,わが国の臨床研究体制を確立する上で大変参考になる。 米国では治療研究を行う場合,施設単位の研究では研究のスピードや情報の伝達が遅いこと,臨床試験データの品質確保,データ管理や生物統計学などの必要性が当初から指摘されていた。 これらを改善するため米国では早くから国家的プロジェクトとして,がん薬物療法の臨床試験を行う多施設共同研究グループを作り, がんに有効な新規抗がん剤の臨床開発を行うとともに,多施設共同の公費臨床試験により治癒を目指した標準的化学療法を確立することが必要であると定められた。 これを推進するため,米国の国立がん研究所(NCI: National Cancer Institute)は1952年Zubrodを臨床センターの所長に迎え,1955年にはClinical Studies Panelを作り, 臨床研究医師が多施設共同研究グループ機構に乗って臨床試験に参加すれば,がんや白血病の研究は急速に進むだろうという結論をまとめた。 早くも1955年に米国NCIに対し,がん化学療法プログラム(Cancer Chemotherapy Program: CCP)が与えられ,1959年度には総額1810万ドル(当時の日本円で約66億円)が予算化された。 米国NCIはうち380万ドル(約14億円)をかけ,1959年までに18の共同研究グループを発足させた4)。 このようにがんに対する共同研究グループが全て国の公的研究費の支援をうけて設立されたことは特記すべきことである。 米国では新薬開発や患者に対する最善の治療法や標準的治療法を確立するための臨床試験研究は国の研究政策の一環として行われたことは注目すべきである。
このために設立された共同研究グループのいくつかを紹介すると,Acute Leukemia Group A (ALGA)やAcute Leukemia Group B (ALGB)が1955年にまず設立された。ALGAは小児の,ALGBは成人の白血病を対象にした共同研究グループであったが,最近では両者が合体し,それに固形がんも対象に加え,グループの名前もCancer and Leukemia Group B (CALGB)に発展し,その研究活動はめざましいものがある。また,ジョンズホプキンス大が中心となって作られたEastern Solid Tumor Group (ESTG)は固形がんを対象にしていた。これは現在のECOG (Eastern Cooperative Oncology Group)へ発展した。1956年にできたSouthwest Cancer Chemotherapy Group (SWCCG)は小児の悪性腫瘍を対象にしていたが,数年後には成人の悪性腫瘍を対象にしたSouthwest Oncology Group(SWOG)へと発展し,現在に至っている。
現在このような大きな共同研究グループは11に整理統合されているが,1955年の当初から各共同研究グループには統計部門(statistical service)をもつように計画され,臨床試験研究に生物統計学の導入とデータ管理業務がきわめて大切であるという認識があったことは,わが国と大きく異なる点である。 また,この臨床試験研究に対し公的研究費が十分支援され,1994年頃の一つの共同研究グループの年間研究費は約20億円,データセンターは約5億円である。 したがって,少なくとも11グループで250億円以上の公的研費が投入されていることになる。1994年のNCIの研究費の予算では,臨床研究に対し4.4億ドル, そのうち臨床試験研究に対しその42%である1.87億ドル(当時の日本円で280億円)が支援されている5)ので,この推定は間違ってはいないことが分かる。
このように公的研究費で支援された共同研究グループができてはじめて優れた新規抗がん剤が多数開発されるようになった。 しかし,いかに優れた新規抗がん剤といえども,単剤ではがんを治すことはできないことも明らかになってきた。小児の急性リンパ性白血病(ALL)を治せる治療法ができたのは,1964年のVAMP療法[vincristine (VCR), methotrexate (MTX), 6 - mercaptopurine, prednisolone(PDN)]で,これがさらに改良され,約40%の症例が治癒する治療法として,VP - cp療法(VCR, PDN, +脳脊髄X線照射)が確立したのは1970年のことである。一方,ホジキン病(HD)に対し,1967年に治癒可能なMOPP療法(mechlorethamine, VCR, procarbazine, PDN)が開発され,現在の標準的治療法であるABVD療法[adriamycin(ADM), bleomycin, vinblastine, dacarbazine)]が1975年に開発された。ランダム化比較試験でこれらの治療法が評価されて,ABVD療法が標準的治療法として確立したのが1992年である。 一方,非ホジキンリンパ腫の標準的治療法であるCHOP療法(cyclophosphamide, ADM, VCR, PDN)は1976年に発表され,それが大規模なランダム化比較試験によって標準的治療法として確立されたのは,1993年である。
このような大規模なランダム化比較試験はごく小さな共同研究グループでは遂行できないため,研究グループの規模を拡大する必要に迫られるようになった。 このため1976年にNCIは多施設共同研究の参加施設を増やすため,共同研究拡大プログラム(CGOP: Cooperative Group Outreach Program)を導入した。これとは別に1983年には地域臨床腫瘍学プログラム(CCOP: Community Clinical Oncology Program)が作られた。CGOPというのは,たとえばSWOGなどの多施設共同研究グループのメンバー施設によって直接監督される研究者が, 研究プロトコールに従って症例登録した場合,第II相試験では1例あたり250ドルまで,第III相試験では500ドルまでの研究費が支払われる研究制度である。 一方,CCOPは地方がんセンターや基幹がんセンターで診療できる研究者が既存の共同研究グループに参加することにより,CCOPの研究費が受けられるようになり, 特定のプロトコール研究に症例登録してもらう制度である6)。このように米国NCIは臨床医に対し臨床研究費を出して公費臨床試験を推進し, 大規模なランダム化比較試験を可能にして,標準的治療法を確立するという制度を作ってきたのである。
米国ではこのような臨床研究体制の中で,企業,国,研究病院,臨床研究医師が一体となって新薬を開発するシステムが作られるとともに, 白血病,悪性リンパ腫,小児がん,固形がんの一部などに有効な新規抗がん剤が数多く開発され,治癒を期待した併用療法や集学的治療法の研究が急速に進んだ。 しかし,治癒が期待できる併用療法,すなわち標準的治療法ができたのは前述したNCI主導の公費臨床試験を行う共同研究グループ機構が適切に稼動してからである。 また,複数の治療法を適正に比較するためには,適切な規模のランダム化比較試験を行えるようになったのはCGOPやCCOPが制度化されてからである。 この結果,米国の多施設共同研究グループは研究規模が拡大し,大規模なランダム化比較試験が行えるようになり,各種の悪性腫瘍に対する標準的治療法が確立した。 治療研究の質を高め研究データの信頼性を確保するために,研究病院にリサーチナースやデータマネージャーなどの研究コーディネーターという職種が導入された。 このような臨床研究体制の下で行われた臨床試験を通してがん治療の質の高い証拠(evidence),すなわち標準的治療法が確立した。個々の患者に対し,この証拠に基づいて最善の医療が行われる仕組み(EBM, evidence-based medicine)のほとんどは米国の多施設共同研究グループによる公費臨床試験により確立されたのである。 これが可能になったのは,国を上げて臨床試験を行うために必要な共同研究グループ機構や臨床研究体制の整備が早くからなされてきたことによる。
わが国にも多施設共同研究グループはあったが,多くは散発的で非継続的なものであり,また統計センターや各種委員会をそろえ, かつ継続して公的研究費の支援を受けた共同研究グループ機構はなく,その体質は極めて貧弱なものであった。われわれは1978年にがん研究助成金による共同研究グループを立ち上げたが, それを発展させて,統計センターや各種委員会をもち,公的研究費で支援された共同研究グループ機構が形成されたのは,1991年のJCOG (Japan Clinical Oncology Group,日本臨床腫瘍研究グループ)が最初と思われる。しかしその公的研究費はSWOGのものに比し10分の1以下である。 全国の共同研究グループに対する公的研究費は人口比で調整してもわが国は米国の100分の1以下であるといっても過言ではない。
わが国には共同研究グループ機構に必須な生物統計の専門家も極めて少なく,臨床研究医師は臨床試験の科学的方法や臨床疫学の教育もされていないため, 研究デザイン,研究プロトコール,CRF,研究規模の設定などは科学的評価に耐えられるものが少なかった。 またデータセンターやデータ管理のシステム,モニタリングや監査といった品質管理や品質保証を実行するシステム,およびそれを担当するリサーチナース,データマネージャーといった研究コーディネーターなどの職員に欠けていた。 さらに臨床試験に登録できる症例集積能力を十分に備えた研究病院は極めて少ない状況にある。 このため,わが国では信頼性が十分に確保された臨床試験研究は極めて少ないという状況が長く続いた。 この原因は基本的には臨床試験研究に対する国の研究政策が欠如していることによっているが,このほかに,大学医学部の業績評価が臨床医学部門においてさえ基礎医学研究に偏っており, 臨床医学研究を軽視してきたことにもその原因がある。また,われわれを含め臨床研究医師の研究熱意や力の不足,臨床試験研究は患者に対する標準的治療法の確立に不可欠であり, 患者に対する最善の医療を提供し,医療の質を向上させるためにも不可欠な臨床研究であるということが十分認識されてこなかったことにも原因がある。 さらに新薬開発治験の不祥事,ヘルシンキ宣言に基づく倫理規範の導入の遅れなどにより社会的な理解が得られていないことにも原因がある。 しかし,このような状況は患者にとって,ひいては国民にとってよいことではない。健康科学や生命科学の成果は大きな経済効果をもつことは明らかなので, 基礎研究と臨床研究は一体として推進できるように政府は研究政策を転換する必要がある。 今後は,これらの原因を一つ一つ取り除く努力を積み重ねて,よい研究成果が出せる共同研究グループ機構と臨床研究体制を作っていく必要がある。
このような問題があるとはいえ,各種悪性腫瘍に対して有効な新規抗がん剤が多数開発されてきた。しかし,すぐれた抗がん剤といえども単剤では進行がんを治すことはできない。 したがって,新しい併用療法や集学的治療法を公費臨床試験による科学的方法で評価し,治癒が期待できる標準的治療法を確立することが求められている。 小児の急性リンパ性白血病はがん薬物療法が成功した代表的な疾患であるが,その研究の歴史的展開をみることにより,臨床試験研究の重要性がよく認識できると思われる。
急性リンパ性白血病治療の進歩
1 抗がん剤の開発に伴う治療成績の向上とその限界(1945〜70年)
小児急性リンパ性白血病(ALL)に対し,1945年から1970年までの間に多種類の有効な抗がん剤が開発された。 図1はそれらの抗がん剤の小児ALLに対する治療成績を示したものである。確かに有効な薬の数が多くなればなるほど延命効果はそれなりに認められるが, 当時では治癒させることができなかったのである。しかし,当時の研究で分かったことは生存期間が向上しても,必ず再発するということであった。 その初再発部位を分析した所,約60%の症例は脳,脊髄,すなわち中枢神経組織(CNS: central nervous system)に初再発しており,この原因として病初から極少数のALL細胞がCNSに浸潤しており,血液脳関門(blood brain barrier)によって薬が局所に到達しないために再発することが発見された。
図1 急性リンパ性白血病の治療の進歩
1.抗がん剤の開発に伴う治療成績の向上とその限界(1946〜70)
2 治癒を目指した集学的治療法の開発と治癒率の向上(1962年〜71年)
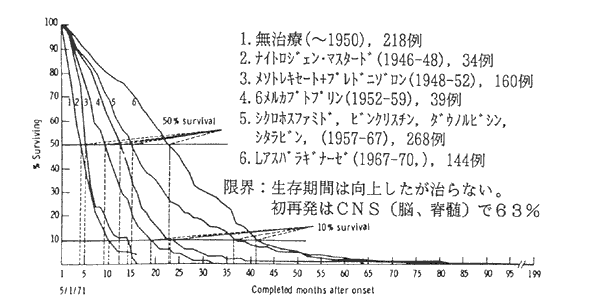
1964年開発されたVAMP療法で治癒率が10%程度に得られるようになったが,CNSに初再発率が60%以上に起こることから,寛解導入療法の初期からCNSに放射線治療を行う臨床試験が開始された。 図2は,その成果を図示したものである。照射線量が5〜12Gyでは効果はないが, 24GyではCNS再発は5%と低下し,生存率がかなり向上することが明らかになった。また,MTXの髄注でも同じくらいの効果があるということも明らかになった。 こういった公費臨床試験研究を積み重ねた結果,現在では小児ALLの約80%は治るという結果が出ている。 これは公費臨床試験による成果であり,新薬の開発治験だけでは達成できないことである。患者に対する最善の治療法や標準的治療法を確立するために, 研究者主導の公費臨床試験を行い,これにより初めて治癒率が向上したということで,よい研究モデルとなっている。
図2 急性リンパ性白血病の治療の進歩(1962〜71)
2. 集学的治療法の開発と治癒率の向上(小児)
SWOGの規模
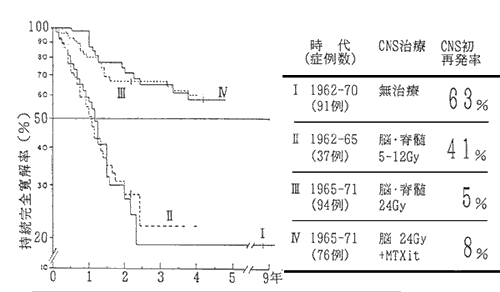
このような臨床試験研究を展開している米国の共同研究グループであるSouthwest Oncology Group (SWOG)の共同研究グループ機構7)を図3に示した。 SWOGの本部はテキサス大の健康科学センターにありそこにグループ代表者がいる。SWOGの統計センターはフレッドハッチンソンがんセンターにあり, この両者が協力してこのグループの臨床試験研究を管理している。
図3 Southwest Oncology Group(SWOG)の機構(1992)
多施設共同研究グループはdisease committeeと呼ばれ,乳がん,胃がん,泌尿器がん,白血病,リンパ腫などの14の臓器がん共同研究グループがある。 さらにdiscipline committeeとして病理,放射線,外科などの委員会があり,それぞれの分野について施設のクオリティコントロールを行っている。 さらに常設の各種委員会をもっている。参加施設の在り方はわが国とかなり異なっている。すなわち,34のメンバー施設(member institution)が指定されており,その施設に所属する診療科の研究責任医師(研究者)はすべてSWOGの研究に参加するということになっている。 一方,わが国では施設単位の参加ではなく,診療科単位に参加する,すなわち,外科が参加しても,内科は参加しないという,診療単位に参加するという状況がある。
SWOGでは,メンバー施設の監督下にCGOPの制度で参加してくる291人の研究者がおり,登録症例数に応じて研究費が支援されている。 これとは別にCCOPの制度によって地方がんセンターや基幹がんセンターで診療できる26人の研究者が参加している。 また泌尿器科がんの共同研究拡大プログラム(UCOP: Urologic Cancer Outreach Program)によって23人の研究者が, さらにNCIが重要と認定したハイプライオリティの臨床試験研究について,26人の研究者が指名されて参加している。 SWOGの研究にかかる費用の95%以上は公的研究費によって支援されている。SWOGの年間の研究費は約20億円である。
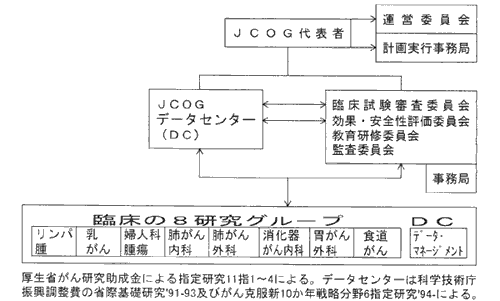
JCOGの研究機構
一方,わが国の現状はどうか,JCOGを例にとり説明したい。JCOGは図4に示すように八つの臓器がん共同研究グループと各種委員会, データセンターからなる共同研究グループ機構をもち,その年間の公的研究費は1.8億円である8)。 JCOGでは,1991年にデータセンターをつくるにあたり,科学技術庁から年間5000万円の研究費を得て,3年間でデータセンターを立ち上げた。 その後はがん克服新10カ年戦略分野6の指定研究班として年間3000万円前後の研究費でデータセンターの開発を続けると同時にJCOGデータセンターとして研究業務を行っている。 一方,SWOGの統計センターは年間費用が5〜6億円であり,SWOGの年間研究費が20億円であるのと比較すると,JCOGはSWOGの約1/10の規模である。
図4 Japan Clinical Oncology Group(JCOG)の研究機構:1999年度
(JJCO 28:158,1998) 一部改訂
(JJCO 28:158,1998) 一部改訂
日米欧の代表的ながん共同研究グループの統計センターの規模の比較
表2はSWOG,EORTC,JCOGの統計センターの規模を比較したものである。 アクティブな試験数や年間登録数で非常に大きな差があるほか,SWOGのデータセンターには統計家, コンピューター専門家,データマネージャー,データエントリー,秘書など約50人の職員がおり,EORTCでは約30人,一方JCOGでは正規の職員はおらず, 非常勤またはアルバイトがせいぜい1〜2人というところである。これはJCOGではその公的研究費の使用に規制があることと,その額が決定的に少ないことによっている。
表2 がん共同研究グループの統計センターの規模

データ管理とデータマネージャー
アメリカでは臨床試験研究を行う場合,NCIやNIHから研究費をとる必要があり,その場合には,その研究に参加している生物統計家やデータマネージャーがいることが必須条件になっている。データマネージャーの業務内容について,Sloan - Ketteringの場合9)では,出身学部は問わないが,Bachelor(学士)の資格をもっていること,さらに6〜12カ月の研修で医療情報の収集,データ入力ができることが要求される。
データ入力とその取扱いには責任をもち,したがってデータマネージャーはパスワードでデータベースを管理しているので,他人がデータの入力変更することはできない。 1人あたり6〜8人の患者を受けもっており,リサーチナースと協力してCRFのデザイン,入力フォームも作成している。研究責任者の指導の下で研究業務を行い,監査や査察にも対応している。
リサーチナースのレベルと業務内容
リサーチナースのレベルと業務内容も,施設によってさまざまであるが,Sloan-Ketteringでは四つのレベルに分けられている9)。
レベル1は正看護婦で2年以上の専門医療分野の臨床経験があり,研究プロトコールの内容,用語を理解し,薬物投与,治療,反応モニター,検体採取等ができる人である。
レベル2では,レベル1を6カ月以上経験して,IRBやGCPおよびGCP関連ガイドライン,薬効別臨床評価ガイドラインを理解していることが条件になる。 適格規準による登録例の判断と決定ができ,インフォームド・コンセントの取得に参加している。もちろん研究責任者の指導下であるが,日本でいえば若手医師の研究業務が行えるというレベルである。
レベル3はレベル2を1年以上の経験があって,教育的指導やプロトコールの評価,論評ができることが要求されており,わが国でいえばほぼ研究分担医師程度の実力がある。
レベル4は日本では不可能なレベルである。すなわちレベル3を1年以上経験し,専門医療分野で扱う特殊な疾患やその医学的知識をもつ人である。 専門看護師,たとえばオンコロジーナースやナースプラクティショナーの資格をもつことが要求される。このナースプラクティショナーはcommon diseaseに対する処方やプロトコールに記載されている処方や検査オーダーができる資格をもっているが,わが国ではこれは制度化されていない。 欧米にはこのようにレベルの高いリサーチナースがいるからこそプロトコール研究を安全で質の高い研究として実施できるわけである。 もちろん研究責任者の指導の下で,これらの研究業務が行われているが,これらはプロトコール研究の信頼性や科学性を保つために必要な職種であるという認識がある。
臨床試験運用上のよいシステム
がんのNSABP(National Surgical Adjuvant Breast and Bowel Project)というグループでは下記の四つのシステムが動いている9)。
1.Nursing guideline:プロトコールごとに使用薬剤の薬理,合併症,ADR(adverse drug reaction)に対する対応,薬剤投与時のモニターなどの指針が書かれている。
2.Toll free Telephone Line for Clinical Questions:現場の施設からの問い合わせに対して連携を緊密にして質問に答えるシステムで,だいたい月に600〜700回の問い合わせがある。
3.Mentor Program:よい指導者のプログラムということであるが,必要に応じて本部よりリサーチナース,データマネージャーを現場に派遣して指導等の支援を行っている。
4.Site Visit Audits:現地へ出向いて訪問監査を行うシステムであるが,監査はあくまで教育的に行われている。 NSABPでは年200回くらいの頻度で行われており,全施設は3年間で必ず訪問監査を受けることになっている。監査委員の費用はすべて実費支給で,謝金は出されていない。監査はピアレビュー的に行われている。
米国の臨床試験の研究費
米国における臨床試験研究では,院内のIRBで承認されたプロトコールとともに必要な研究費の申請書を添付し,たとえば米国NIH(National Institute of Health)やNCI等へ研究費の申請をする必要がある。
研究費は直接経費と間接経費に分かれている。直接経費は研究に必要なkey personell(リサーチナースとデータマネージャー)の 人件費と試験薬や検査費用などの研究用実費である。人件費の積算は共同研究医師であれば,10万ドル/年,研究生(医師)5万ドル/年, リサーチナース5〜6万ドル/年,データマネージャー4〜5万ドル/年,薬剤師は1投与あたり30ドルの基準をもとにして,週50時間として,その% effortを算出して積算される9)。
間接経費はgrantが得られたら,追加して支払われる研究実施施設の必要経費であり,主として施設の共通人件費,運営経費,備品経費などが相当する。 NIHのgrantでは,これらの経費としてgrantの50%が追加支給され,その分は施設が研究を遂行する上で必要となる共通人件費や運営費として使うことができる。 治験の場合では研究費の35%,その他の基金ではその10〜15%が間接経費として追加支給される9)。 したがって,公費臨床試験のgrantがとれれば,施設の機能が充実するということになる。 わが国ではこの間接経費の制度はなく,国立の施設で治験を行うと研究費の30%が技術料等の名目で国庫に収納され,施設にはおりてこないというおかしな制度になっているのも問題である。
がん臨床試験の研究体制の構築に必要なもの:政策の日米比較
わが国と米国になぜこのような差ができたかということが問題である。 表3に示すようにこれには両国間で健康科学・生命科学・医療技術研究の研究政策に大きな相違がある。 たとえばアメリカでは政府によるNIHの研究費は最近で年間1.3〜1.4兆円ぐらい出ている。 一方,わが国では1995年はその1/10の約1000億円である。1998年になり,公的研究費は2倍以上増えて2500億円になった。 しかし,ほとんどは基礎研究費として使われており,臨床研究費,特に臨床試験のための研究費はその1/100も支援されていないと思われる。 一方,米国では1994年度でみるとがん研究に対し,1.4兆円のうちの約25%である約3000億円がNCIの研究費となり,うちがんの臨床研究にその21%(630億円)が, うち約280億円ががん共同研究グループの公費臨床試験に対し支援されている。 つまり,わが国のがん共同研究グループに対する研究費は米国に比し1/100以下であることはこの点よりも明らかである。
研究を総括する機関は日本では文部省,科学技術庁,厚生省の縦割りであるが,アメリカではNIHが一元管理をしているため,基礎から臨床までうまく流れることになる。
研究政策自体は米国の大統領教書にあるように,「科学は経済成長のエンジンであり,技術はその燃料」という認識がであって,医学・生命科学の研究はどの領域よりも一番重要視されている。 要するに医学生命科学の研究は経済成長に使っているわけで,遺伝子研究はしたがって全部パテントをとるという姿勢である。遺伝子研究も単に新しい遺伝子をみつけるというのみならず, その作用や臨床における有用性を明確にするという基礎から臨床へ向けて一貫した研究政策を構築している。 遺伝子研究そのものは良いことなのか,悪いことなのかという議論があるが,そのことは別にして,こういった研究政策の違いがあることを認識しておく必要がある。
もう一つは,アメリカではヒトを対象にしたすべての研究に対して国家研究法の規制がかけられている。したがって治験も公費臨床試験もともにこれがかかっている。 規制をする代わり研究費も出すということである。日本はやっと厚生省の私的諮問機関の厚生科学会議が科学技術基本法を作ったり,研究費の増額をはかってきたが, これらはすべて基礎研究が対象で,臨床研究を対象としていないのは大変残念なことである。
臨床研究の倫理的な規制についても,アメリカの薬事法(FDC)は1962年に抜本改正があり,臨床研究に対しインフォームド・コンセント(IC)を必須化することは1966年から入ってきている。 臨床研究に対するICやIRBなどの制度は1974年の国家研究法で定められ,その結果臨床研究を行う場合,倫理委員会やIRBがない施設には研究費を出さないというNIHの指令が出ている。 また,遺伝子技術に対してもNIHはRAC(recombinant DNA advisory committee)やHGT(human gene therapy, point to consider)の指針を作り, 1981年には早くもGCPが完成したのである。つまり,建築でいえば1階を固めて2階,3階を積み上げていくという手法で研究政策は倫理面を含め手堅く立ち上げてきた。 一方,わが国では臨床研究に対する研究政策はなく,倫理的な規制もかけてこなかった。 しかし国際情勢の影響があり,1989年より治験のみに対し,旧GCPの規制をかけ,1998年より本格的な新GCPの規制をかけてきたが,公費臨床試験に対しては, このような規制もかけず,また研究費も支援しないという状況が続いている。遺伝子技術の審査は文部省と厚生技術の縦割りに分かれ, 最近になりやっと施設の認可だけでよいことになったという状況である。つまりきわめて対症療法的な制度作りであり,例えていえば, 1階を作らないで2階を作っているという状況が続き,したがって臨床試験でいえば共同研究グループ機構も作らなければ,臨床研究の研究体制も作られてこなかった。 そのため治験や臨床試験は空洞化するのは当然のなりゆきである。
ま と め
これをいったいどう改善したらよいかということである。一つはやはり臨床研究体制を確立することである。 治験を公務にしたために医療職の医師は定員も増えないまま診療と臨床研究を行うことになるので,その負担は大きい。 臨床研究の重要性を認識し,適正な臨床研究ができる研究体制と研究をする医師の定員枠を作るべきである。 そしてCRCを定員化することが必要である。2点目は臨床研究全体に国家研究法で規制をかけ,その代わりきちんと制度化するべきである。 公費臨床試験や臨床研究に対する倫理規制,特に国家研究法の制定が必要で, すべての臨床試験研究に対し,ICH-GCPやICHガイドラインが適応されるように,研究政策の方針を転換することが必要である。 そうしないと,企業による新薬開発治験は制度化し重要視するが患者に対する最善の治療法や標準的治療法を確立するために必要な医師研究者主導による公費臨床試験を制度化しない, つまり,それは重要視しないという事になり,わが国の政府や行政の研究政策が批判されることになる。 第3は臨床研究,特に臨床試験研究に対する公的研究費の支援をそれなりに行うことが必要である。 第4は臨床試験研究に必須な共同研究グループ機構をきちんと作ることである。そのための公的研究費を支援すべきである。
文 献
1) ICH関係通知集’98,およびその追補.薬事時報社;1998.および改訂GCPハンドブック.薬事時報社;1997.
2) 国立病院・療養所における治験等受託研究の取扱い.株式会社 ミクス;1999.
3) 厚生省通知,研第4号/医薬審第104号.適応外使用に係る医療用医薬品の取扱いについて.1999.
4) The national program of cancer chemotherapy research.Cancer Chemother Rep 1959;5 - 41.
5) Funding for clinical (patient - oriented) oncology research.Current status and recommendations for improvement. J Clin Oncol 1996;14:666 - 70.
6) Frelick RW.The community clinical oncology program(CCOP) story:review of community oncologists' experiences with clinical research trials in cancer with an emphasis on the CCOP of the National Cancer Institute between 1982 and 1987.J Clin Oncol 1994;12:1718 - 23.
7) Sowthwest Oncology Group Statistical Center.1997.
8) Shimoyama M, et al.Japan Clinical Oncology Group(JCOG).Jpn J Clin Oncol 1998;28:158 - 62.
9) 山田博豊,下山正徳.米国における臨床試験実施体制の現状と日本における今後の臨床試験の在り方:がん臨床試験研究体制の在り方取材調査報告.臨床医薬 1998;16:2807 - 20.