■JPT-online■
| 薬理と治療(JPT)vol.30 no.7 | |
 |
|
| ◆ 第1章 序論─本邦の臨床試験の仕組み─ | |
| ◆ 第2章 医療機関の特性と臨床試験の実施状況 | ◇ 第1節 目的と背景 ◇ 第2節 方法 ◇ 第3節 結果 ◇ 第4節 考察 |
| ◆ 文 献 | |
|
|
|
第1章 序論─本邦の臨床試験の仕組み─
医薬品の開発は,複数のプレイヤーが参加・貢献することにより成し遂げられるある種の社会的事業である。 その開発過程では,当該物質の有する好ましい性質や好ましくない性質に関する情報がさまざまな手法により収集される。
医薬品開発に関する方法論は,医薬品を巡る規制環境および医療現場の実状の変化を踏まえ,時代とともに発展しつつある 11)Pocock SJ. Clinical Trials. New York: John Wiley & Sons;1983.14-27. ~2)藤井基之.創薬論.東京:薬事日報;1995.7-32.3)3)Lesko LJ, Rowland M, Peck CG, Blaschke TF. Optimizing the science of drug development: opportunities for better candidate selection and accelerated evaluation in humans. J Clin Pharmacol 2000;40:803-14.。 特に近年,遺伝子レベルの探索により病因タンパク質を同定し,これを標的として作用するようデザインされた化合物を選択し開発する手法(いわゆるゲノム創薬)の 応用に向けての動きには目覚ましいものがある。 このような手法の導入により,たとえば,効果発現や副作用発現の個人差,ばらつきを予見し,また,コントロールすることが可能になると考えられる。
見出された医薬品候補物質について,動物実験において安全性が確認され,薬理学的に十分な作用を示した場合には, その毒性プロファイルも考慮しつつ,ヒト(最初は,通常,健康成人)においてその安全性, 場合によっては有効性に結びつく可能性のある薬理学的作用の発現が確認される第1相試験が実施される。 医薬品の開発過程のうち,臨床試験とはこの段階以降を指す。
臨床試験という言葉は,薬剤(被験薬),医療用具等,さらには外科的手法や介護等も含め, これらをヒトに適用したときの評価を行うための科学的な実験一般に対して用いられる。 一方,治験という用語は,薬事法においては特別の意味合いで定義されている。
薬事法における治験とは,医薬品等の承認取得を目的とする試験(薬事法第2条第7項)である。 すなわち,厚生労働省に対して,将来,医薬品等の製造承認または輸入承認を申請するためのデータを収集する臨床試験が治験である。 なお,医薬品等とは医薬品と医療用具を指すが,本論文では特に断らない限り,医薬品に焦点を当てた議論を展開する。 治験は,その意味では,将来の承認申請を目的意識として有する者(通常は製薬企業)の存在により規定される概念である。
本邦での臨床試験,特に薬事法で規定される治験が現在どのような体制で実施されているかをFig.1-1に示す。 この仕組みは,直接には,薬事法の下位の命令である「医薬品の臨床試験の実施に関する基準」により規定されている 4)4)医薬品の臨床試験の実施の基準に関する省令. 平成9年3月27日厚生省令第28号.。 この「医薬品の臨床試験の実施に関する基準」,いわゆるGood Clinical Practice(以下「新GCP」と略す)は,国際ハーモナイゼーション会議( International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use,以下「ICH」と略す)における 日,米,欧三極の合意に基づき,平成9年(1997年)4月に日本に導入され,平成10年(1998年)4月より全面施行されたルールで, 欧米の臨床試験のルールをその基礎とする。
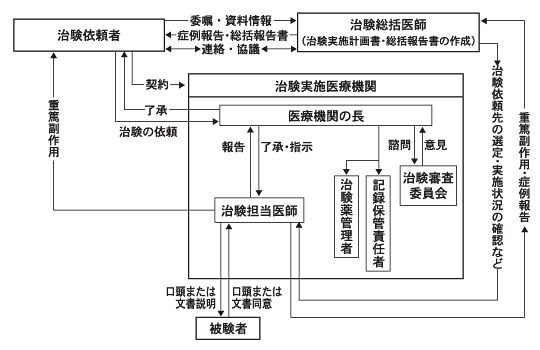
なお本邦においては,このICH-GCP導入以前にも(平成2年以来), ICH-GCPよりもはるかに緩やかなGCP(このGCPを以下「旧GCP」と呼ぶ 5)5)医薬品の臨床試験の実施に関する基準について. 平成元年10月2日薬発第874号. が臨床試験のルールとして存在していたが,薬事法に直接の根拠を有するものではなく,実施上の限界があった。 Fig.1-2に旧GCPに基づく臨床試験の実施体制を示す。
Fig.1-2に示す旧GCP下の臨床試験の特徴は,治験総括医師が試験の実施主体となっている点である。 治験総括医師は,プロトコルの立案,施設の選択から,最終的な総括報告書の作成に至るまでの責任を一手に担うとされていた。 多くの場合,個々の試験分野の重鎮とされる大学教授らがその任を引き受けていたのだが,実際上の問題として, 多忙を極めるそのような総括医師が臨床試験実施の事実上の牽引車となりうるのかについては疑問視されることもあった。 たとえば,試験の最終成果である総括報告書の作成にあたって,「案」を製薬企業が作成し, それを総括医師が確認し署名するという慣行が当然のようにあったが,かかる慣行は試験の実施・結果に対する責任の所在を不明確にしていた。
Fig.1-1に示す新GCPの下では,日本的な「たてまえ」で成り立っていたそのような仕組みが, より現実的なものに置き換えられた6)6)小野俊介.改正GCPの要点.月刊薬事 1997;39(10):65-7.。 その特徴としてもっとも重要な点は,プロトコルの立案と総括報告書の作成が治験依頼者の責務として明確に位置付けられたことである。 かつての総括医師は,医学専門家として外部から製薬企業にアドバイスを行うこととなった。 また,試験が医療機関で適切に実施されていることを治験依頼者である製薬企業が確認するための仕組み(モニタリング,監査)が 実施体制の中に義務として組み込まれている。医療機関の側にもモニタリング,監査を受け入れる義務があるとされる。
被験者(臨床試験に参加する患者や健康人)の権利・健康を守るための仕組みも強化された。 インフォームドコンセントを得るプロセスが整備され,より詳細な情報が文書で被験者に伝わるようになった。 また,医療機関ごとに設置が義務付けられている治験審査委員会(Institutional Review Board,以下「IRB」という)による事前の審査プロセスも, 旧GCP下に比べて強化されている。
このように,臨床試験を実施する仕組みは,欧米のシステムを日本の医療システムに一見うまく組み込んだものとなった。 しかし,注意すべきは,臨床試験の仕組みの世界共通化は,臨床試験の目的や開発における位置づけ, 社会における臨床試験の価値の共通化を必ずしも意味しない点である。 すなわち,本邦の試験には,実施医師,製薬企業,被験者を取り巻く社会・医療環境を反映して,欧米とは異なる評価方法が採用され, また,特定の試験形態が好まれる等の特徴がある。
新医薬品の承認の取得を目的とした臨床試験は,多くの場合,私的企業の長期的な利潤追求のための新薬開発の一段階であるが, その実施を一つの社会的事業と捉えた場合,公的分野の直接的な厳しい規制の下で実施されている点が一般的な私的事業との大きな相違点である。 すなわち,臨床試験がどのように実施されているか,そして,臨床試験がどうある「べき」かを論じるには, 関係者のモチベーションおよび関係者に対するインセンティブが歴史的背景に基づく公的規制あるいは支援策にどう影響されているのかという観点が必要である。
施策としての平成9年(1997年)の新GCPの導入は,結果として規制環境の劇的な変化となった。 この変化に対応して,関係者はおのおの合目的的な選択を行い,試験に参加(あるいは不参加)しているが, 果たしてその結果は「好ましい」と考えられるものであったのかが政策評価の観点からの問題である。
この問題に答えることは容易ではない。たとえば,「好ましい」かどうかの判断の基準を何に求めるのか, 観点としては社会全体からのそれだけで良いのか,「好ましい」の判断基準との関係において,どのような分配的正義を妥当として採用するのか, 代替案との関係で実現可能性はどうか等の公共政策論における重要かつ難解な諸問題がいくつもある。
さらに,臨床試験に関する政策を論じる際には,実務的な問題として,入手可能な情報の質・量が極めて限定されていることも前提とする必要がある。 臨床試験の実施状況は,製薬企業にとって秘匿性のある情報であり,通常は公開情報として得られることはない。 近年,十分な数の被験者を確保するために新聞等で試験の実施が広告されているが,具体的な医療機関に関する情報はそこには盛り込まれない 7)7)斉藤宏暢.被験者募集に係わるアンケート調査.ミクス Feb.2001:53-8.。 厚生労働省に提出が義務付けられている治験届の情報も,特に激しい開発競争が行われている分野における競争への影響, 情報公開法における対応との一貫性を考えると,現時点では,医療機関の情報を含む詳細なデータが開示されることはないと考えられる。
本研究には大きく二つの目的がある。一つは,上述した困難,制約の中で臨床試験に関する政策の評価を行うための前提となる基本データを分析し, 整理して提示することである(第2章,第3章)。もう一つの目的は,政策評価を行う際に必要な視点について論じ, 一般的に適用可能で理解しやすい考え方の枠組みを提示することである(第4章)。 これらはいずれも本分野の政策評価における必須の論点であるにもかかわらず, たとえば,本邦において新GCPが導入された際に十分な議論が行われたとは考えにくい点である。
政策評価に関して,政府は必要性,効率性,公平性といった観点から具体的な評価を行うこととしており, そのガイドラインも平成13年(2001年)に公表されている8)8)政策評価各府省連絡会議.政策評価に関する標準的ガイドライン.平成13年1月15日.。 本研究の結果は,現実の医薬品政策を評価するに際しての評価のエンドポイント,視点,機会費用の考え方等を示す一つの道標となりうるものと考える。
なお,本研究において示す論考は,基本的に,近年幅広く用いられている医療経済学の発想に基づく。 その意味で著者は,治験を実施する医師,医療機関,患者,規制当局たる厚生労働省のいずれの立場をもa prioriに肯定, あるいは,否定するものでない。国の機関が社会厚生の最大化のみをその行動原理にしているのではないことは, これらの機関の「合理的」行動に関する歴史的な研究が明らかにしている。また,特に倫理を論ずる場合には, 経済学的な考え方自体に興味を示さない立場,あるいはそれを積極的に否定する立場もあることには注意が必要である。
Fig.1-1 新GCP下の治験実施体制
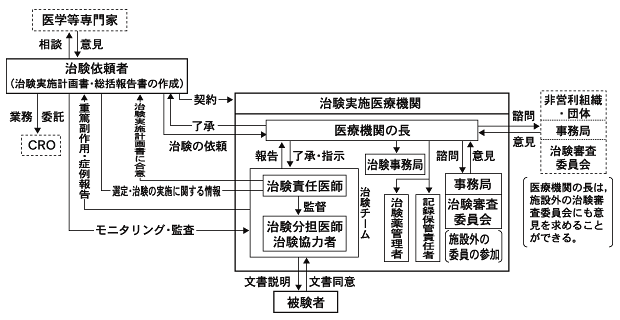
Fig.1-2 旧GCP下の治験実施体制